Craniopharyngioma
Craniopharyngioma is a rare, non-cancerous (benign) brain tumor that originates near the pituitary gland and hypothalamus, critical areas that regulate many bodily functions.

Key Takeaways
- Craniopharyngioma is a rare, benign brain tumor typically arising from Rathke’s pouch remnants near the pituitary gland.
- Symptoms often include vision problems, headaches, and hormone deficiencies due to pressure on surrounding structures.
- Diagnosis involves neurological examination, MRI scans, and endocrine function tests.
- Treatment primarily consists of surgical removal, often followed by radiation therapy to manage residual tumor.
- Long-term management frequently requires hormone replacement therapy and regular monitoring due to the tumor’s location and potential for recurrence.
What is Craniopharyngioma: Definition and Causes
To understand what is Craniopharyngioma, it’s essential to recognize it as a rare type of brain tumor that develops from remnants of Rathke’s pouch, an embryonic structure involved in pituitary gland development. These tumors are typically benign, meaning they are not cancerous and do not spread to other parts of the body. However, their location near vital brain structures, such as the optic nerves, pituitary gland, and hypothalamus, can lead to significant health issues. The craniopharyngioma definition and causes highlight its origin and the impact it can have on endocrine function and vision.
Craniopharyngiomas are relatively uncommon, accounting for approximately 1-5% of all primary brain tumors. Their incidence is estimated at 0.5 to 2.0 cases per million people per year, according to the National Organization for Rare Disorders (NORD).
What Causes Craniopharyngioma?
The exact cause of craniopharyngioma is not fully understood. It is believed to arise from residual embryonic cells of Rathke’s pouch that fail to fully regress during fetal development. While most cases are sporadic, some research suggests a link to specific genetic mutations, particularly in the CTNNB1 gene, which plays a role in cell growth and development. However, these genetic changes are not typically inherited, meaning the tumor does not usually run in families. Environmental factors have not been definitively linked to the development of craniopharyngiomas.
Craniopharyngioma Symptoms and Diagnosis
The symptoms of craniopharyngioma vary widely depending on the tumor’s size, location, and its impact on surrounding brain structures. Early recognition of craniopharyngioma symptoms and diagnosis is crucial for effective management. Symptoms often arise from pressure on the optic nerves, hypothalamus, or pituitary gland, leading to visual disturbances, hormonal imbalances, and neurological issues.
Common Symptoms of Craniopharyngioma
Patients with craniopharyngioma may experience a range of symptoms, which can develop slowly over time:
- Vision problems: Often the most common symptom, including blurred vision, loss of peripheral vision (bitemporal hemianopsia), or double vision, due to compression of the optic chiasm.
- Headaches: Persistent or worsening headaches, sometimes accompanied by nausea and vomiting, resulting from increased intracranial pressure.
- Hormonal deficiencies: The tumor can disrupt the pituitary gland’s ability to produce hormones, leading to symptoms such as:
- Growth failure in children (due to growth hormone deficiency).
- Delayed puberty or menstrual irregularities.
- Increased thirst and urination (diabetes insipidus).
- Fatigue, weight gain, or sensitivity to cold (hypothyroidism).
- Neurological symptoms: Changes in personality, memory problems, confusion, or balance issues, especially if the tumor affects the hypothalamus.
Diagnosing Craniopharyngioma
Diagnosis typically begins with a thorough neurological examination and a review of symptoms. The diagnostic process for craniopharyngioma involves several key steps:
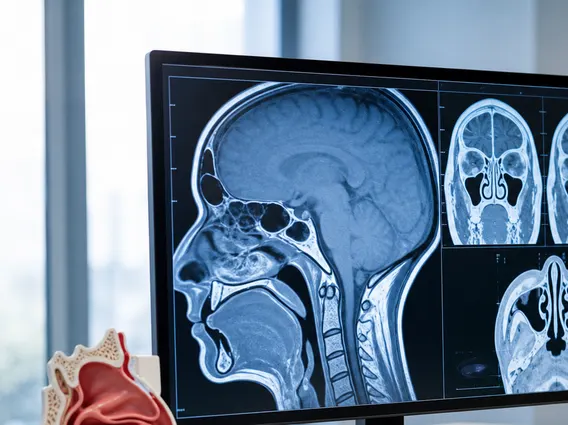
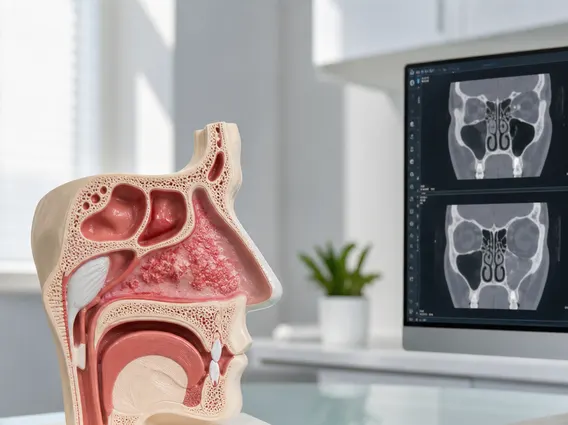
- Imaging studies: Magnetic Resonance Imaging (MRI) of the brain is the primary diagnostic tool, providing detailed images of the tumor’s size, location, and relationship to surrounding structures. Computed Tomography (CT) scans may also be used, particularly to detect calcifications within the tumor, a common feature of craniopharyngiomas.
- Endocrine evaluation: Blood tests are performed to assess pituitary hormone levels (e.g., growth hormone, thyroid hormones, cortisol, prolactin, sex hormones) to identify any deficiencies.
- Ophthalmological examination: A comprehensive eye exam, including visual field testing, is crucial to assess the extent of vision loss.
Craniopharyngioma Treatment and Outlook
Managing craniopharyngioma requires a multidisciplinary approach involving neurosurgeons, endocrinologists, radiation oncologists, and ophthalmologists. The primary goal of craniopharyngioma treatment options explained is to remove as much of the tumor as safely possible while preserving neurological and endocrine function. The craniopharyngioma prognosis and outlook depend on several factors, including the tumor’s size, location, and the extent of surgical removal.
Available Treatment Options
Treatment strategies are individualized based on the patient’s age, tumor characteristics, and overall health:
- Surgery: This is the cornerstone of treatment. The aim is to achieve maximal safe resection.
- Transsphenoidal surgery: A minimally invasive approach through the nose and sphenoid sinus, often used for tumors primarily within the sella turcica.
- Transcranial surgery (craniotomy): An open surgical approach through the skull, used for larger or more complex tumors.
- Radiation therapy: Often used after surgery, especially if there is residual tumor or if complete surgical removal is not possible. Types include:
- Proton beam therapy: Delivers highly targeted radiation, minimizing damage to surrounding healthy tissue.
- Intensity-modulated radiation therapy (IMRT): Uses advanced technology to shape radiation beams to the tumor’s contours.
- Stereotactic radiosurgery: A highly focused form of radiation that delivers a high dose to the tumor in one or a few sessions.
- Cyst aspiration: For cystic craniopharyngiomas, fluid can be drained to relieve pressure, sometimes with intracystic chemotherapy or radiation.
Long-Term Prognosis
The craniopharyngioma prognosis and outlook are generally favorable for survival, as these tumors are benign. However, due to their critical location, patients often face long-term challenges related to hormonal deficiencies, visual impairment, and hypothalamic dysfunction. Lifelong follow-up care is essential, including:
- Hormone replacement therapy: Most patients will require lifelong hormone replacement for deficiencies in growth hormone, thyroid hormone, cortisol, or sex hormones.
- Regular monitoring: Periodic MRI scans are necessary to monitor for tumor recurrence or growth of residual tumor.
- Management of hypothalamic dysfunction: This can include addressing issues like obesity, sleep disturbances, and behavioral changes.
While the tumor itself is benign, its treatment and long-term effects can significantly impact quality of life, necessitating ongoing medical support and management.